„Fenomenologikus termodinamika” változatai közötti eltérés
(→Fázisdiagramok) |
|||
| 186. sor: | 186. sor: | ||
===Fázisdiagramok=== | ===Fázisdiagramok=== | ||
| − | + | [[Fájl:151-2_a víz fázisdiagramja.jpg|bélyegkép|jobbra|300px|A víz fázisdiagramja]] | |
| + | [[Fájl:Titan_nikkel_fazisdiagram.jpg|bélyegkép|balra|300px|A titán-nikkel ötvözet fázisdiagramja]] | ||
==Kémiai potenciál== | ==Kémiai potenciál== | ||
A lap 2011. június 20., 16:44-kori változata
A tétel korábbi változata megtekinthető a Fenomenologikus termodinamika (2010) címszó alatt. Kevésbé struktúrált, de részletesebb, mint az új.
Tartalomjegyzék
Termodinamikai állapotjelzők
Az állapotjelző a termodinamikai rendszernek egy olyan jellemzője, amely csak a rendszer állapotától függ, és nem függ attól, hogyan jutott a rendszer ebbe az állapotba. Az állapotjelző a rendszer egyensúlyi állapotát írja le. Például a belső energia, az entalpia, entrópia, nyomás és hőmérséklet állapotjelzők, mivel kvantitatíve jellemzik egy termodinamikai rendszer egyensúlyi állapotát. Ugyanakkor a mechanikai munka és a hő nem állapotjelző, mivel kvantitatíve a termodinamikai rendszerek egyensúlyi állapotai közötti átmeneteket írják le. Megkülönböztetünk intenzív és extenzív állapotjelzőket.
Hőtágulás
Hőtágulásnak nevezzük azt a fizikai jelenséget, amikor valamely anyag a hőmérsékletének változásával megváltoztatja a méretét. Melegítéskor az anyagok általában tágulnak, a tágulás relatív mértékét a hőtágulási együttható fejezi ki. A hőtágulás általában közelítőleg lineárisan függ a hőmérséklettől, ez alól kivétel, ha halmazállapot-változás történik, illetve néhány speciális, vagy bomlékony anyag zsugorodik (negatív hőtágulás). Léteznek kerámiák és fémötvözetek, amelyek gyakorlatilag nem változtatják a méretüket.
A lineáris hőtágulási együttható a szilárd anyag hőmérséklet változásra adott hosszméret változásának a mértéke:
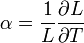 [K-1]
[K-1]
A térfogati hőtágulási együttható az anyagok termodinamikai tulajdonsága, melyet az alábbi összefüggéssel definiálnak[1]:
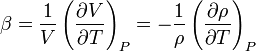 [K-1]
[K-1]
ahol 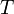 a hőmérséklet,
a hőmérséklet, 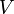 a térfogat,
a térfogat, 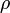 a sűrűség, a deriválást állandó nyomás mellett hajtják végre; β pedig a sűrűség változásának mértéke állandó nyomáson, a hőmérsékletváltozás hatására.
a sűrűség, a deriválást állandó nyomás mellett hajtják végre; β pedig a sűrűség változásának mértéke állandó nyomáson, a hőmérsékletváltozás hatására.
Ideális gáz, kinetikus modell
A gázok törvényszerűségei leírhatók a mozgó testekre vonatkozó fizikai törvényekkel, ha feltételezzük ideális voltukat, amihez a következő kritériumoknak kell teljesülniük:
- A gázmolekulák saját térfogata elhanyagolható a gáz által betöltött térfogathoz képest
- A gázmolekulák egymásra sem vonzó, sem taszító hatást nem fejtenek ki, az ütközésektől eltekintve
- A gázmolekulák egymással illetve az edény falával való ütközése rugalmas
- A gázmolekulák átlagos sebességét és kinetikai energiáját a gáz hőmérséklete adja meg
- Azonos hőmérsékleten, azonos számú gázmolekula kinetikai energiája megegyezik, és független a gáz anyagi minőségétől
Az ideális gázokra, és csak az ideális gázokra teljesül az egyesített gáztörvény.
Általában számításoknál a gázokat – első közelítésben – ideális gázoknak tekintjük. A légnemű közegek jellemzően akkor közelítik meg a tökéletes gázokra jellemző tulajdonságokat, ha hőmérsékletük kritikus hőmérsékletüknél nagyobb (ahol a párolgáshő nulla). Azokat a légnemű anyagokat, amelyeknek hőmérséklete a kritikus hőmérséklet alatti, gőznek hívjuk.
Nyílt és zárt folyamatok
Egy zárt termodinamikai rendszer a különböző állapotait meghatározott valószínűséggel veszi fel. A lehetséges állapotok összességét jellemzi az állapothalmaz:
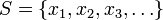 .
.
Az állapotok termodinamikai valószínűsége (hagyományosan)
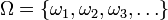 .
.
A rendszer entrópiája az 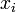 állapotban
állapotban
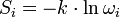 ,
,
ahol k a Boltzmann-állandó. A termodinamikai entrópia fogalmát Clausius vezette be.
Az entrópianövekedés és entrópiamaximum elve
Ha egy rendszer adiabatikusan zárt (vagyis a környezetéből nem vesz fel hőt), akkor a rendszerben lejátszódó spontán folyamatok során a rendszer entrópiája mindaddig nő, amíg be nem áll az egyensúlyi állapot. Egyensúlyi állapotban a rendszer entrópiája maximális.[2] Azonban nyílt rendszer egyensúlyának nem feltétele az entrópiamaximum, mivel az entrópianövekedés a külvilágnak leadott hővel kompenzálható, sőt, az entrópia akár csökkenhet is.
Carnot-folyamat
A Carnot körfolyamat, ha hőerőgépként működik, négy állapotváltozásból áll:


- A gáz reverzibilis izoterm tágulása (expanziója) a TH[3] nagyobb hőmérsékleten (izoterm hőközlés). Ez alatt az állapotváltozás alatt (Az 1. ábrán A állapotból B állapotba) a táguló gáz munkát végez a dugattyún. A gáz tágulását a nagy hőmérsékletű tartályból beáramló hő okozza.
- Izentrópikus (reverzibilis adiabatikus) tágulás. Ennél az állapotváltozásnál (B-ből C-be) feltesszük, hogy a henger és a dugattyú hőszigetelt: nem kap, és nem is veszít hőt a rendszer. A gáz tovább tágul, munkát végezve a környezetén. Ennek eredményeképp a gáz a hidegebb TC[4] hőmérsékletre hűl.
- Reverzibilis izotermikus összenyomódás (sűrítés, kompresszió) a TC hideg hőmérsékleten (izoterm hőleadás). (C-ből D-be). Ekkor a környezet végez munkát a gázon, miközben hő áramlik a gázból a hideg tartályba.
- A gáz izentrópikus összenyomódása. (D-ből A-ba) Ismét felételezzük, hogy a dugattyú és a henger hőszigetelt. A környezet végez munkát a gázon miközben összenyomja azt, ezáltal a hőmérsékletét TH-ra emelve. Az állapotváltozás végén a gáz a kiindulási állapotba jut vissza.
Főtételek
0. főtétel: a termodinamikai rendszer egyensúlya
A nulladik főtétel tulajdonképpen nem egyetlen "törvényt", hanem több posztulátumot jelent, amelyek a termodinamikai rendszer egyensúlyával kapcsolatosak. Ezek:
- bármely magára hagyott termodinamikai rendszer egy idő után egyensúlyi állapotba kerül amelyből önmagától nem mozdulhat ki;
- egy egyensúlyban levő termodinamikai rendszer szabadságfokainak száma a környezetével megvalósítható kölcsönhatások számával egyenlő;
- a két testből álló magára hagyott termodinamikai rendszer egyensúlyban van, ha a testek között fellépő kölcsönhatásokat jellemző intenzív állapothatározóik egyenlők;
- az egyensúly tranzitív (ha A rendszer termodinamikai egyensúlyban van C rendszerrel és B rendszer is termodinamikai egyensúlyban van C rendszerrel, akkor ebből következik, hogy A és B rendszer is termodinamikai egyensúlyban van egymással).
I. főtétel – Energiamegmaradás törvénye
A termodinamika első főtétele mennyiségi összefüggést állapít meg a mechanikai munka, a cserélt hő és a belső energia változása között.
Egy nyugvó és zárt termodinamikai rendszer belső energiáját, amennyiben annak belsejében nem zajlik le fázisátalakulás vagy kémiai reakció, kétféleképpen lehet megváltoztatni: munkavégzéssel és hőközléssel. A rendszer 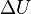 belső energiájának megváltozása tehát a vele közölt Q hőmennyiség és a rajta végzett W (bármilyen) munka összege:
belső energiájának megváltozása tehát a vele közölt Q hőmennyiség és a rajta végzett W (bármilyen) munka összege:
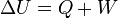
Áramló közegre a hő és a technikai munka összege így számolható:
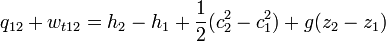
ahol q a hő, w(t12) a technikai munka, h az entalpia, c a közegáramlás sebessége, g a gravitációs állandó, és z a vizsgált pont magassága (helyzete). Differenciális alakban:
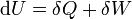
Következménye: Nincs olyan periodikusan működő gép, ú.n. elsőfajú perpetuum mobile (örökmozgó), mely hőfelvétel nélkül képes lenne munkát végezni.
II. főtétel
A második főtétel a spontán folyamatok irányát szabja meg. Több, látszólag lényegesen különböző megfogalmazása van.
- Clausius-féle megfogalmazás (1850.): A természetben nincs olyan folyamat, amelyben a hő önként, külső munkavégzés nélkül hidegebb testről melegebbre menne át. Csakis fordított irányú folyamatok lehetségesek.
- Kelvin-Planck-féle megfogalmazás (1851., 1903.): A természetben nincs olyan folyamat, amelynek során egy test hőt veszít, és ez a hő munkává alakulna át. Szemléletesen egy hajó lehetne ilyen, amelyik a tenger vizéből hőenergiát von el és a kivont hőenergiával hajtja magát. Ez nem mond ellent az energiamegmaradásnak, mégsem kivitelezhető.
Az ilyen gépet másodfajú perpetuum mobilének nevezzük, tehát az állítás szerint nem létezik másodfajú perpetuum-mobile.
A két megfogalmazás egymásból következik, de a levezetése nem teljesen egyszerű.
A második alaptörvénynek ezek és az ezekhez hasonló megfogalmazásai zavarbaejtőek, hiszen a fizika többi, összefüggéseket megállapító törvényeivel szemben valaminek a létezését tagadják. Egy jobb megfogalmazás végett egy új fogalom került bevezetésre: az entrópia. A termodinamika második alaptörvénye az entrópia felhasználásával a következőképpen fogalmazható meg: a spontán folyamatok esetében a magukra hagyott rendszerek entrópiája csak növekedhet.
III. főtétel
Nernst megfogalmazása szerint az abszolút tiszta kristályos anyagok entrópiája nulla kelvin hőmérsékleten zérus. Olyan abszolút tiszta kristályos anyag, amelyre a Nernst megfogalmazása érvényes lenne, a természetben nem fordul elő, ideális fogalom, tehát nulla entrópiájú anyag nem létezhet. Az entrópia határértékét a harmadik főtétel pontosított megfogalmazása a következőképpen rögzíti: a termodinamikai rendszerek entrópiája véges pozitív érték felé, az entrópia hőmérséklet szerinti deriváltja pedig a zéró felé tart, amikor a rendszer hőmérséklete az abszolút nulla érték felé közelít. Nernst posztulátumát később egy újabb megfogalmazásban hozta nyilvánosságra, mely szerint az abszolút nulla hőmérséklet tetszőlegesen megközelíthető, de nem érhető el. E kijelentés a harmadik főtétel előbbi megfogalmazásának következménye: mivel az abszolút nullához közeli hőmérsékleten az anyagok fajhője nagyon kicsi, igen kis hőmennyiség a hőmérséklet jelentős megváltozásához vezet. Bármilyen módon is valósítjuk meg a hűtést, a lehűtendő test valamilyen fokú visszamelegedése elkerülhetetlen. A folyamat megismétlésével a hőmérséklet tovább csökkenthető, tehát végső soron az abszolút nulla hőmérséklet elvileg tetszőleges pontossággal aszimptotikusan megközelíthető, de nem érhető el.
Termodinamikai potenciálok
Bizonyos termodinamikai paramétereket elterjedten neveznek termodinamikai potenciál-függvényeknek is. Ez annyit jelent, hogy - hasonlóan a mechanikában és az elektrodinamikában értelmezett potenciál-függvényekhez - skálázásuk egy állandó erejéig önkényesen végezhető el, azonban a választott skálától függetlenül szélsőértékük helye egyértelműen kijelöli a vizsgált rendszer egyensúlyi állapotát.
Ötféle termodinamikai potenciált különböztetünk meg[5]:
| Név | Jelölés | Képlet | Természetes változók |
|---|---|---|---|
| Belső energia | 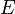
|
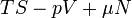
|
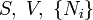
|
| Helmholtz szabadenergia | 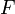
|
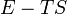
|
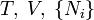
|
| Entalpia | 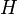
|
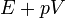
|
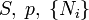
|
| Gibbs szabadentalpia | 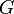
|
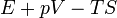
|
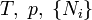
|
| Landau potenciál | 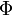
|
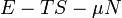
|
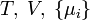
|
A fundamentális egyenlet
Fundamentális egyenletnek azt nevezzük, ami három extenzív változótól függ, pl: S(N, E, V). Fundamentális egyenletből a makroszkopikus rendszerre vonatkozó információk mind kinyerhetőek (ha ezt tudjuk, a rendszer egész termodinamikáját ismerjük).
A fundamentális egyenlet (S(E, V, N)) differenciális alakja:
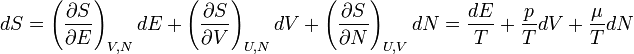
Megfelelően alkalmazva Legendre-transzformációkat a többi potenciál differenciális alakja:
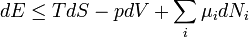
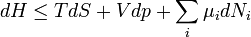
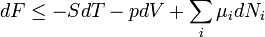
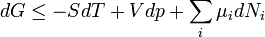
Van der Waals gázok

A létező (reális) gázok tulajdonságai többé-kevésbé eltérnek az ideális gázok tulajdonságaitól. Az eltérés oka abból adódik egyrészt, hogy a gázatomok, -molekulák kölcsönösen vonzzák egymást – ún. van der Waals-erők működnek közöttük –, másrészt nem pontszerűek, van kiterjedésük, azaz saját térfogattal rendelkeznek.
Ha az ideális gáz egyenletébe beírjuk a nyomás- és térfogati korrekciókat, akkor n anyagmennyiség esetén a:
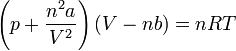
kifejezést kapjuk, amely a reális gázokra vonatkozó van der Waals-egyenlet.
Ha az összefüggésből kifejezzük a nyomást, az alábbi, viszonylag bonyolult összefüggéshez jutunk:
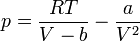
Elvégezve a kijelölt műveleteket és rendezve az egyenletet, a
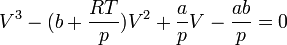
kifejezésből látható, hogy a térfogatra nézve a függvény harmadfokú, mint a mellékelt ábra – a szén-dioxid p–V diagramjának – izotermáin is látható.
Reális gázokkal végezhető kísérletek pl. Gay-Lussac, Joule-Thomson
Fázisátalakulások
- a régi verzióban: Fázisátalakulások termodinamikai tárgyalása és Fázisegyensúlyok
Jellemzői
A termodinamika által leírt folyamatok közül kiemelkedő fontosságúak azok, ahol a folyamat kezdeti anyaga és a folyamat végén keletkezett anyag lényegesen különböző tulajdonságokkal rendelkezik.[6]
A fázisátalakulásokat gyakran kíséri a túlhűtés, ritkábban a túlhevítés jelensége. Előbbit használja ki a Wilson-féle ködkamra, utóbbit a buborékkamra.
Típusai
Elsőrendű fázisátalakulás
Elsőrendű fázisátalakulásnak olyan folyamatot hívunk, amikor az intenzív paraméter folytonos változása mellet az extenzív paraméternek ugrása van. A fázisátalakuás kifejezésen rendszerint elsőrendű fázisátalakulást értünk.
Másodrendű fázisátalakulás
Másodrendű fázisátalakulás az elsőrendűtől abban különbözik, hogy itt az extenzív paraméter is folytonos, csak a deriváltjában van rendkívüli viselkedés. Ilyenek például a kritikus opaleszcencia, ferromágneses átalakulás a Curie-hőmérsékleten, ferroelektromos katasztrófa, szupravezetés, szuperfolyékonyság, stb.
Gibbs-féle fázisszabály
Egy termodinamikai rendszer egyensúlyi állapotában egyszerre jelen levő fázisok (fizikailag homogén részek) száma (F), a szabadon választható állapothatározók (vagy szabadsági fokok) száma (Sz) és a rendszert alkotó független komponensek száma (K) közötti összefüggés:
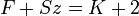
A fázistörvény a termodinamika főtételeiből vezethető le. Pl. ha a víz (egykomponensű rendszer, K = 1) csak egy fázisban (pl. gőzként) van jelen (F = 1), akkor nyomása és hőmérséklete (bizonyos határokon belül) tetszőlegesen változtatható (Sz = 2); míg három különböző fázisban (gőz, folyékony víz és jég, F = 3) már csak egyetlen, meghatározott nyomás- és hőmérsékletértéknél (hármaspont) lehet (Sz = 0).
Fázisdiagramok


Kémiai potenciál
Fázisegyensúlyok
Jegyzetek, hivatkozások
- ↑ Donald L. Turcotte, Gerald Schubert: Geodynamics, 2nd Edition, Cambridge, 2002, ISBN 0-521-66624-4
- ↑ Holics László: Fizika összefoglaló, Typotex Kiadó, 1998, 335. old., ISBN 963-9132-13-6
- ↑ A H jelölés az angol „hot”, azaz „forró”, „meleg” szót rövidíti
- ↑ A C jelölés az angol „cold”, azaz „hideg” szót rövidíti
- ↑ Alberty (2001) p1353
- ↑ Tichy Géza, Kojnok József: Hőtan, Typotex Kiadó, 2001, 75. old., ISBN 963-9326-14-3
- ↑ J. W. Gibbs, 1878