A statisztikus fizikai szimulációk alapjai és a Monte Carlo módszer
Tartalomjegyzék
Statisztikus fizikai szimulációk alapjai
Legyen egy sokaságunk, aminek az energiája 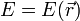 , ahol
, ahol 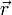 az összes szabadsái fokot tartalmazza (mechanikai rendszerre
az összes szabadsái fokot tartalmazza (mechanikai rendszerre 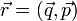 ). Ha a rendszer követi a Boltzmann-statisztikát, akkor a rendszer
). Ha a rendszer követi a Boltzmann-statisztikát, akkor a rendszer ![[\vec{r}, \vec{r}+d \vec{r}]](/images/math/5/6/1/56187ee1de8ce46a085984701d48d250.png) intervallumban való megtalálási valószínűsége:
intervallumban való megtalálási valószínűsége:
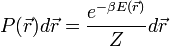
Egy A mennyiség átlagát a sokaságon a következőképp számoljuk (PS = phase space = fázistér):
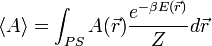
Az integrált csak akkor tudjuk egzaktul kiszámolni, ha a fázistér minden pontjára kiértékeljük a függvényt, és úgy átlagolunk. A Monte-Carlo integrálással ezt a problémát oldhatjuk meg. Az 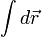 helyére
helyére 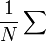 írunk, így egy véges összeget kell elvégeznünk. A pontok számát (N) az határozza meg, hogy mekkora pontossággal akarjuk az integrált meghatározni. Az integrál dimenziójától függetlenül a hiba
írunk, így egy véges összeget kell elvégeznünk. A pontok számát (N) az határozza meg, hogy mekkora pontossággal akarjuk az integrált meghatározni. Az integrál dimenziójától függetlenül a hiba 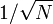 szerint csökken, vagyis 1% pontossághoz 10000 pontot kell számolni.
szerint csökken, vagyis 1% pontossághoz 10000 pontot kell számolni.
Molekuladinamika
A molekuladinamika a részecskék mikroszkopikus dinamikájának követésével foglalkozik. Valódi rendszerben 1023 nagyságrendű részecske van, ezt a mai számítógépekkel szimulálni lehetetlen. Azonban ennél kevesebb részecskét is elég szimulálnunk ahhoz, hogy a termodinamikai tulajdonságokat vizsgálhassuk. A szimulációkban a párkölcsönhatásokat vesszük figyelembe, ám lehetséges közelítéseket tenni. A részecskék közt a Van der Waals erő hat, ami elég gyorsan lecseng, így távoli részecskék közt elhanyagolható (ezen a pontot különbözik a molekuladinamika és a gravitációs N-test szimuláció, ahol az 1/r2-es erő miatt nem hanyagolható el a kölcsönhatás).
A szimulációban van tehát N darab részecske, melyekre a Newton-törvények alapján kiszámítjuk a rájuk ható erőt, majd léptetjük a rendszert. Mivel a kezdőállapotokat általában nem a termodinamikai egyensúlyból indítjuk, meg kell várni, hogy a rendszer beálljon abba. Hogy mikor állt be, azt az ekvipartíció segítségével mutathatjuk ki:
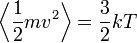
Ha a rendszer elérte az egyensúlyi állapotát, mérhetővé válnak a termodinamikai változók (hőmérséklet, nyomás, hőkapacitás, stb.)
Fizikai mennyiségek mérése a szimulációkban
Pillanatnyi hőmérséklet
Bár azt mondtuk, hogy a rendszert az egyensúlyi állapotában vizsgáljuk, a nem egyensúlyi állapotban is bevezethetünk egy pillanatnyi hőmérsékletnek nevezett mennyiséget. Termikus egyensúlyban igaz az ekvipartíció:
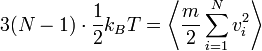 ,
,
ahol 3(N-1) a szabadsági fokok száma (a TKP 3 koordinátája van levonva). A nem egyensúlyi helyzetben, bár nem igaz az ekvipartíció, ezt a képletet alkalmazhatjuk a hőmérséklet meghatározására.
Teljes energia
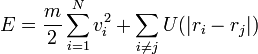
Hőkapacitás
Fluktuáció-disszipáció tétel alapján:
![C_V = \frac{1}{k_BT^2}[\langle E^2 \rangle - \langle E \rangle^2]](/images/math/5/6/6/5665d1b98f0b1a2a48c050809050fe1e.png)
Nyomás
A nyomást a viriál-tétel segítségével fejezhetjük ki:
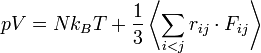
Verlet-algoritmus
Molekuladinamikai szimulációkban legtöbbször a Verlet-algoritmust használják a diffegyenletek megoldására. A Runge-Kuttával szemben több előnye van:
- gyorsabb, mert egy lépésben csak egyszer kell a gyorsulásokat számolni
- majdnem olyan pontos, mint a RK4 (
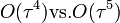 )
) - jól megőrzi az energiát
- időtükrözésre nem változik (ez a részleges egyensúly feltétele miatt fontos)
A Verlet-algoritmus egy lépése (R(t) a koordináták, V(t) a sebességek, A(t) a gyorsulások):
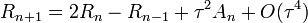
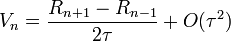
Hátrányai:
- két előző lépést használ, így nem indítható 1 kezdeti feltételből
- a sebesség és a pozíció nem egyszerre frissítődik, a sebesség "le van maradva"
Megoldás: velocity-Verlet algoritmus:
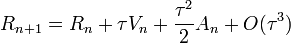
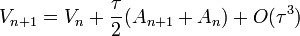
- R-ben már csak
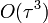 pontosságú
pontosságú - ez már nem 2 előző lépést használ
- koordináták és sebességek egyszerre frissülnek
- sebesség is pontosságú
A Metropolis algoritmus
A Metropolis algoritmus egy eljárás, amivel bonyolult valószínűségi eloszlások mintavételezhetőek. Másképp megfogalmazva szeretnénk előállítani egy 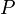 eloszlást, a kérdés az, hogy milyen átmeneti valószínűségekkel
eloszlást, a kérdés az, hogy milyen átmeneti valószínűségekkel 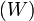 vezérelt folyamat (egészen pontosan Markov-folyamat) vezet ilyenre? A módszer kihasználja a részletes egyensúly elvét, ez esetünkben azt jelenti, hogy:
vezérelt folyamat (egészen pontosan Markov-folyamat) vezet ilyenre? A módszer kihasználja a részletes egyensúly elvét, ez esetünkben azt jelenti, hogy:
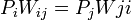 teljesül minden
teljesül minden 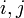 -re.
-re.
Ez még kevés feltétel az átmeneti valószínűségekre, ezért a Metropolisz megkötés a következő:
![W_{ij} = \mathrm{min}\left[1, \frac{P_j}{P_i} \right]](/images/math/a/d/1/ad1920c41b7e81ab4a74e9500c031123.png)
Bár általánosan tetszőleges eloszlás előállítható, fizikában általában valamilyen egyensúlyi eloszlást szeretnénk előállítani. Például, ha termális egyensúlyt szeretnénk, akkor 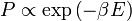 , és a Metropolisz megkötésben szereplő hányados is exponenciális alakban írható. A -t előállító eljárás a következő:
, és a Metropolisz megkötésben szereplő hányados is exponenciális alakban írható. A -t előállító eljárás a következő:
- Induljunk ki egy A konfigurációból.
- Állítsuk elő a megváltoztatott B konfigurációt.
- Számoljuk ki erre a két konfigurációra a megkötésben szereplő hányadost.
- Generáljunk egy 0-1 intervallumon vett egyenletes eloszlású véletlen számot: X.
- Ha
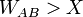 akkor a B konfigurációt fogadjuk el, ellenkező esetben marad az A.
akkor a B konfigurációt fogadjuk el, ellenkező esetben marad az A. - Ismételjük 1-től.
A Monte-Carlo módszer
A Monte-Carlo módszernek nevezzük az olyan eljárásokat, amelyek a problémákat random számok és valószínűségek felhasználásával oldják meg. Az eljárás során ismétlődően kiértékelünk egy determinisztikus modellt, random számokat használva inputnak. Akkor használják, ha a feladat nagyon összetett, nemlineáris, vagy nagyon sok paramétertől függ.
Használata:
- Állítsuk föl a modellt: y = f(x1, x2, ..., xq)
- Generáljunk random számokat inputnak: xi1, xi2, ..., xiq
- Értékeljük ki a modellt, az eredményt tároljuk el yi-ben
- Ismételjük a 2. és 3. lépéseket n-szer
- Elemezzük az eredményeket hisztogram, összesítő statisztikák, stb. segítségével