„A statisztikus fizikai szimulációk alapjai és a Monte Carlo módszer” változatai közötti eltérés
| 1. sor: | 1. sor: | ||
== Statisztikus fizikai szimulációk alapjai == | == Statisztikus fizikai szimulációk alapjai == | ||
== Molekuladinamika == | == Molekuladinamika == | ||
| + | A molekuladinamika a részecskék mikroszkopikus dinamikájának követésével foglalkozik. Valódi rendszerben 10<sup>23</sup> nagyságrendű részecske van, ezt a mai számítógépekkel szimulálni lehetetlen. Azonban ennél kevesebb részecskét is elég szimulálnunk ahhoz, hogy a termodinamikai tulajdonságokat vizsgálhassuk. A szimulációkban a párkölcsönhatásokat vesszük figyelembe, ám lehetséges közelítéseket tenni. A részecskék közt a Van der Waals erő hat, ami elég gyorsan lecseng, így távoli részecskék közt elhanyagolható (ezen a pontot különbözik a molekuladinamika és a gravitációs N-test szimuláció, ahol az 1/r<sup>2</sup>-es erő miatt nem hanyagolható el a kölcsönhatás). | ||
| + | |||
| + | A szimulációban van tehát N darab részecske, melyekre a Newton-törvények alapján kiszámítjuk a rájuk ható erőt, majd léptetjük a rendszert. Mivel a kezdőállapotokat általában nem a termodinamikai egyensúlyból indítjuk, meg kell várni, hogy a rendszer beálljon abba. Hogy mikor állt be, azt az ekvipartíció segítségével mutathatjuk ki: | ||
| + | |||
| + | <math>\left \langle \frac{1}{2}mv^2 \right \rangle = \frac{3}{2}kT</math> | ||
| + | |||
| + | Ha a rendszer elérte az egyensúlyi állapotát, mérhetővé válnak a termodinamikai változók (hőmérséklet, nyomás, hőkapacitás, stb.) | ||
| + | |||
| + | ===Verlet-algoritmus=== | ||
| + | Molekuladinamikai szimulációkban legtöbbször a Verlet-algoritmust használják a diffegyenletek megoldására. A Runge-Kuttával szemben több előnye van: | ||
| + | * gyorsabb, mert egy lépésben csak egyszer kell a gyorsulásokat számolni | ||
| + | * majdnem olyan pontos, mint a RK4 (<math>O(\tau^4) \text{vs.} O(\tau^5)</math>) | ||
| + | * jól megőrzi az energiát | ||
| + | * időtükrözésre nem változik (ez a részleges egyensúly feltétele miatt fontos) | ||
| + | |||
| + | A Verlet-algoritmus egy lépése ('''R(t)''' a koordináták, '''V(t)''' a sebességek, '''A(t)''' a gyorsulások): | ||
| + | |||
| + | <math>R_{n+1} = 2R_n - R_{n-1} + \tau^2 A_n + O(\tau^4)\,</math> | ||
| + | |||
| + | <math>V_n = \frac{R_{n+1} - R_{n-1}}{2\tau} + O(\tau^2)</math> | ||
| + | |||
| + | Hátrányai: | ||
| + | * két előző lépést használ, így nem indítható 1 kezdeti feltételből | ||
| + | * a sebesség és a pozíció nem egyszerre frissítődik, a sebesség "le van maradva" | ||
| + | |||
| + | Megoldás: velocity-Verlet algoritmus: | ||
| + | * R-ben már csak <math>O(\tau^3)</math> pontosságú | ||
| + | * ez már nem 2 előző lépést használ | ||
| + | * koordináták és sebességek egyszerre frissülnek | ||
| + | * sebesség is <math>O(\tau^3)</math> pontosságú | ||
| + | |||
== A Metropolis algoritmus == | == A Metropolis algoritmus == | ||
A Metropolis algoritmussal a statisztikus fizikai rendszer energiaminimumát találhatjuk meg, ahogy azt a [http://mafihe.hu/~wiki/wiki/index.php/Numerikus_m%C3%B3dszerek#Szimul.C3.A1lt_h.C5.91kezel.C3.A9s szimulált hőkezelés] témakörében is láttuk. Az algoritmus a következő: | A Metropolis algoritmussal a statisztikus fizikai rendszer energiaminimumát találhatjuk meg, ahogy azt a [http://mafihe.hu/~wiki/wiki/index.php/Numerikus_m%C3%B3dszerek#Szimul.C3.A1lt_h.C5.91kezel.C3.A9s szimulált hőkezelés] témakörében is láttuk. Az algoritmus a következő: | ||
A lap 2011. június 11., 13:49-kori változata
Tartalomjegyzék
Statisztikus fizikai szimulációk alapjai
Molekuladinamika
A molekuladinamika a részecskék mikroszkopikus dinamikájának követésével foglalkozik. Valódi rendszerben 1023 nagyságrendű részecske van, ezt a mai számítógépekkel szimulálni lehetetlen. Azonban ennél kevesebb részecskét is elég szimulálnunk ahhoz, hogy a termodinamikai tulajdonságokat vizsgálhassuk. A szimulációkban a párkölcsönhatásokat vesszük figyelembe, ám lehetséges közelítéseket tenni. A részecskék közt a Van der Waals erő hat, ami elég gyorsan lecseng, így távoli részecskék közt elhanyagolható (ezen a pontot különbözik a molekuladinamika és a gravitációs N-test szimuláció, ahol az 1/r2-es erő miatt nem hanyagolható el a kölcsönhatás).
A szimulációban van tehát N darab részecske, melyekre a Newton-törvények alapján kiszámítjuk a rájuk ható erőt, majd léptetjük a rendszert. Mivel a kezdőállapotokat általában nem a termodinamikai egyensúlyból indítjuk, meg kell várni, hogy a rendszer beálljon abba. Hogy mikor állt be, azt az ekvipartíció segítségével mutathatjuk ki:
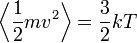
Ha a rendszer elérte az egyensúlyi állapotát, mérhetővé válnak a termodinamikai változók (hőmérséklet, nyomás, hőkapacitás, stb.)
Verlet-algoritmus
Molekuladinamikai szimulációkban legtöbbször a Verlet-algoritmust használják a diffegyenletek megoldására. A Runge-Kuttával szemben több előnye van:
- gyorsabb, mert egy lépésben csak egyszer kell a gyorsulásokat számolni
- majdnem olyan pontos, mint a RK4 (
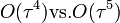 )
) - jól megőrzi az energiát
- időtükrözésre nem változik (ez a részleges egyensúly feltétele miatt fontos)
A Verlet-algoritmus egy lépése (R(t) a koordináták, V(t) a sebességek, A(t) a gyorsulások):
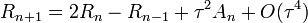
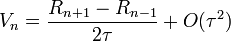
Hátrányai:
- két előző lépést használ, így nem indítható 1 kezdeti feltételből
- a sebesség és a pozíció nem egyszerre frissítődik, a sebesség "le van maradva"
Megoldás: velocity-Verlet algoritmus:
- R-ben már csak
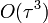 pontosságú
pontosságú - ez már nem 2 előző lépést használ
- koordináták és sebességek egyszerre frissülnek
- sebesség is pontosságú
A Metropolis algoritmus
A Metropolis algoritmussal a statisztikus fizikai rendszer energiaminimumát találhatjuk meg, ahogy azt a szimulált hőkezelés témakörében is láttuk. Az algoritmus a következő:
- Induljunk ki egy A konfigurációból, aminek tudjuk az EA energiáját.
- Változtassunk a rendszeren, hogy egy A-hoz közeli B konfigurációt kapjunk. Számoljuk ki a konfiguráció EB energiáját.
- Ha EB < EA, fogadjuk el ezt az új konfigurációt (így a Boltzmann-faktornak is eleget teszünk).
- Ha EB > EA, az új állapotot
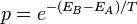 valószínűséggel elfogadjuk.
valószínűséggel elfogadjuk.
A hőmérséklet folyamatos csökkentésével az algoritmus bekonvergál az energiaminimumba.
A Monte-Carlo módszer
A Monte-Carlo módszernek nevezzük az olyan eljárásokat, amelyek a problémákat random számok és valószínűségek felhasználásával oldják meg. Az eljárás során ismétlődően kiértékelünk egy determinisztikus modellt, random számokat használva inputnak. Akkor használják, ha a feladat nagyon összetett, nemlineáris, vagy nagyon sok paramétertől függ.
Használata:
- Állítsuk föl a modellt: y = f(x1, x2, ..., xq)
- Generáljunk random számokat inputnak: xi1, xi2, ..., xiq
- Értékeljük ki a modellt, az eredményt tároljuk el yi-ben
- Ismételjük a 2. és 3. lépéseket n-szer
- Elemezzük az eredményeket hisztogram, összesítő statisztikák, stb. segítségével